 |
|
IMP Manual
develop.4eee3cf66f,2026/08/01
|
To understand the function of a macromolecular assembly, we must know the structure of its components and the interactions between them. However, direct experimental determination of such a structure is generally rather difficult. While multiple methods do exist for structure determination, each has a drawback. For example, crystals suitable for X-ray crystallography cannot always be produced, especially for large assemblies of multiple components. Cryo-electron microscopy (cryo-EM), on the other hand, can be used to study large assemblies, but it is generally limited to worse than atomic resolution. Finally, proteomics techniques, such as yeast two-hybrid and mass spectrometry, yield information about the interactions between proteins, but not the positions of these proteins within the assembly or the structures of the proteins themselves.
One approach to solve the structures of proteins and their assemblies is by "integrative" or "hybrid" modeling, in which information from different methods is considered simultaneously during the modeling procedure. The approach is briefly outlined here for clarity; it has been covered in greater detail previously. These methods can include:
The integrative approach has several advantages:
Hybrid structures based on our integrative approach:
The integrative modeling procedure used here is shown below.
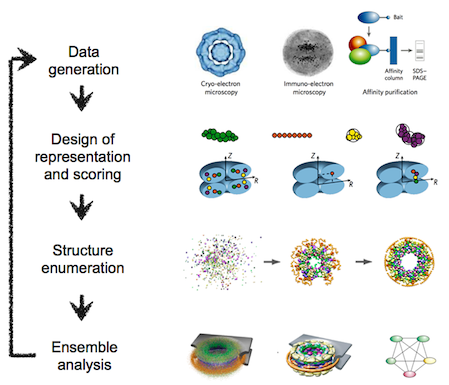
The first step in the procedure is to collect all experimental, statistical, and physical information that describes the system of interest.
A suitable representation for the system is then chosen and the available information is translated to a set of spatial restraints on the components of the system. For example, in the case of characterizing the molecular architecture of the nuclear pore complex (NPC), atomic structures of the protein subunits were not available, but the approximate size and shape of each protein was known, so each protein was represented as a ‘string’ of connected spheres consistent with the protein size and shape. A simple distance between two proteins can be restrained by a harmonic function of the distance, while the fit of a model into a 3D cryo-EM density map can be restrained by the cross-correlation between the map and the computed density of the model.
Next, the spatial restraints are summed into a single scoring function that can be sampled using a variety of optimizers, such as conjugate gradients, molecular dynamics, Monte Carlo, and inference-based methods. This sampling generates an ensemble of models that are as consistent with the input information as possible.
In the final step, the ensemble is analyzed to determine, for example, whether all of the restraints have been satisfied or certain subsets of data conflict with others. The analysis may generate a consensus model, such as the probability density for the location of each subunit in the assembly, and yield a measure of the uncertainty in the solutions.


