 |
The Nup84 complex structure determined in the previous section is consistent with all input information, but for a detailed understanding of its function, an accurate atomic structure is required. Two possible routes to such a structure, depending on the available information, are (i) fitting atomic structures of the individual protein subunits into a cryo-EM map of the assembly and (ii) accurately placing pairs of subunits relative to each other using X-ray crystallography or molecular docking. For both routes, atomic structures of the subunits are required; these structures can be obtained via X-ray crystallography or comparative modeling.
One component of the Nup84 complex is the Nup133 protein; the structure of this protein has been characterized by both X-ray crystallography and small angle X-ray (SAXS). SAXS differs from X-ray crystallography in that it is applied to proteins in solution rather than crystals; thus, it can be applied to a much wider range of proteins in states more closely resembling their functional forms than X-ray crystallography, but the information is rotationally averaged and so the resulting SAXS profile gives less structural information. IMP contains a method that, given an atomic protein structure, can calculate its SAXS profile using the Debye formula, and then fit this profile against the experimentally determined one. This method is implemented in the IMP saxs module and so can be used by writing a suitable Python script. However, because fitting against a SAXS profile is a common task, we provide an IMP application, FoXS, which automates this process. FoXS is available both as a command-line IMP application and a web service.
All input files for this demonstration are available in this zipfile. The structure of the C-terminal domain of yeast Nup133 is available in the RCSB Protein Data Bank (PDB) as code 3kfo (file 3KFO.pdb), while the experimental SAXS profile is given in the 23922_merge.dat file. The atomic structure can be fit against the SAXS profile by running FoXS in the directory containing both files:
foxs 3KFO.pdb 23922_merge.dat
Alternatively, the two files can be submitted to the FoXS web server. FoXS compares the theoretical profile of the provided structure (solid line in the image below) with the experimental profile (points), and calculates the quality of the fit, χ, with smaller values corresponding to closer fits:
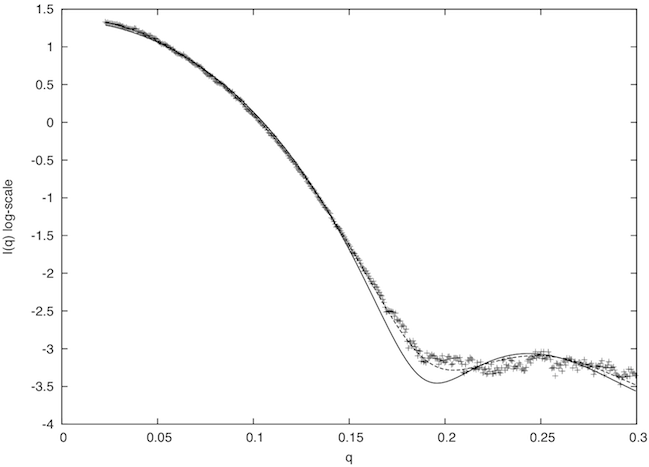
The fit in this example is not a good one (χ=2.96). To understand why this is so, we examine the header of the 3kfo PDB file, which reveals two problems. Several residues at the N and C termini were not resolved in the X-ray experiment (8 in total, 2 at the N terminus and 6 at the C terminus), and the sidechains of 16 other residues could also not be located (REMARK 465 and REMARK 470 lines).
The missing 8 residues and 16 sidechains need to be placed to create a complete atomic structure. One way to achieve this goal is to build a comparative model using a package such as MODELLER relying on the original 3kfo structure as a template and the full sequence (including the 8 missing N and C terminal residues) as the target. The corresponding MODELLER alignment file (3KFO-fill.ali) and script file (fill.py) are provided in the downloaded zipfile. Each candidate comparative model can be fitted against the SAXS profile using the FoXS command-line application or the web service in exactly the same way as the original 3kfo structure; the best MODELLER model gives a significantly improved fit between the theoretical and experimental profiles (dashed line in the above plot; χ=1.21).
Given similar atomic structures of the subunits in the Nup84 complex, as either crystal structures or comparative models, restrainer can be used to build an atomic model of the complex. Note, however, that an accurate model of such a complex would require additional information beyond the proteomics data used above, since yeast two-hybrid data only show that proteins interact, not the specific residues in the protein-protein interaction, and thus do not inform us about the relative orientations of the interacting proteins. Such information can be obtained, for example, from chemical-crosslinking, molecular docking, or cryo-EM maps, as illustrated in the next section.