 |
The IMP C++/Python library offers a great deal of flexibility in setting up the system and restraints. However, in many cases, a simpler interface to solve modeling problems is preferable. The restrainer IMP module is one such interface that simplifies the set up of a complex system, generating the system representation and restraints from a pair of XML files. Optimization, however, may still need to be adjusted for specific cases.
As a simple demonstration of the module, we consider the construction of a model of a subcomplex of the Nuclear Pore Complex (NPC). The yeast NPC is a large assembly of 50 MDa containing 456 proteins of 30 different types. The modeling of the entire assembly is beyond the scope of this tutorial; however, it has been observed that the NPC is made up of a set of smaller subcomplexes, one of which is the Nup84 complex, consisting of seven proteins:
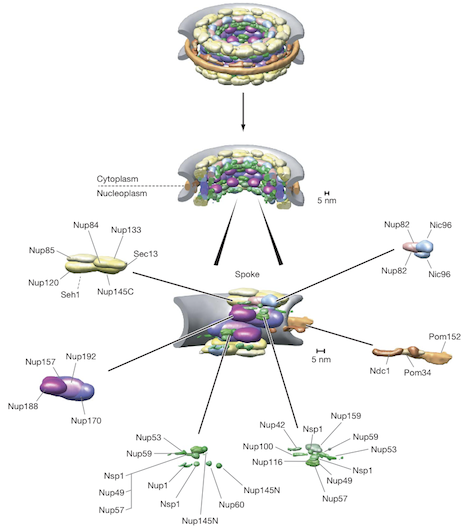
In this tutorial, we will build a model of the Nup84 complex; all of the XML and Python files necessary to perform the modeling can be found in this zipfile. The first of these XML files is representation.xml, which determines how the system is represented. IMP does not require every protein in the system to be modeled with the same representation; for example, some proteins could be modeled as sets of atoms and others at a lower resolution. As for the original NPC modeling, here we use a ‘bead model’ for the Nup84 complex; each protein is represented as a sphere, or a pair of spheres (in the case of the more rodlike Nup133 and Nup120 proteins), with larger proteins using larger spheres. The second XML file encodes the input structural data as spatial restraints on the system. Here, we use two simple sources of information. First, excluded volume for each protein. Second, yeast two-hybrid results for some pairs of proteins. The third XML file is for visualization only, and assigns each sphere a different color. Finally, the Python script loads in all three of the XML files and performs a simple conjugate gradients optimization. This Python script can be executed just like any other Python script:
python nup84.py
restrainer first generates a set of sphere-like particles to represent the system. It then converts the information in the restraints file into a set of IMP restraints. It generates an excluded volume restraint that prevents each protein sphere from penetrating any other sphere and a set of ‘connectivity’ restraints that force the protein particles to reproduce the interactions implied by the yeast two-hybrid experiments. The optimization generates a file optimized.py that is an input file for the molecular visualization program Chimera; when loaded into Chimera, it displays the final optimized configuration of the complex:
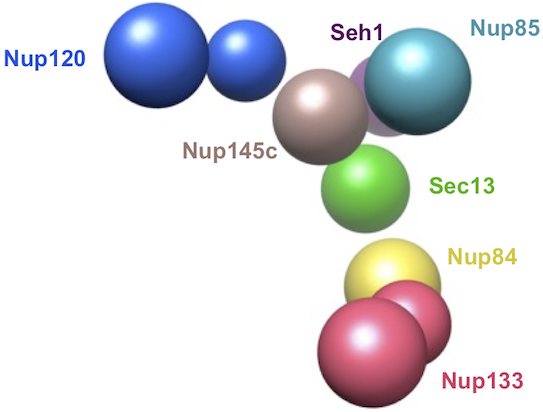
In this example, the modeling problem is simple and thus generating a single model is sufficient to find a solution that satisfies all restraints. However, when all such models need to be found or, in more complex cases, when a global solution of the scoring function is hard to find (for example, because restraints are contradictory due to errors in experiments or experiment interpretations), the modeling procedure is repeated to generate an ensemble of models. When modeling the NPC, the top-scoring models were clustered and used to generate a probability density for each component within the complex. The envelope of this density defined the precision of the corresponding component localization. Only a single cluster of structures was found that satisfied all of the restraints. If contradictory information is presented, however, the optimization will be frustrated, unable to find solutions that simultaneously satisfy all restraints. The ensemble of solutions will exhibit more variability than that in a non-frustrated case. Such frustration can be tested for in the iterative integrative modeling procedure by removing potentially conflicting restraints and repeating the modeling. Finally, the accuracy of the generated model(s) can be gauged by comparison with experimental data that were not used in the original modeling. For example, the generated bead model of the Nup84 complex has a characteristic Y-shape, which is consistent with electron micrographs of the complex, even though these data were not used in our example.
The restrainer XML and Python files, together with the experimental data, such as cryo-EM maps, constitute a complete modeling protocol. Thus, an assembly model built using this protocol can be published along with the input files to allow the model to be reproduced and easily updated. Such a model can thus act as a reference for future studies; for example, regions of the model that were poorly resolved can be investigated with new experiments, the resulting data incorporated into the protocol, and new models generated. Alternatively, existing unused experimental data can be added to the protocol to determine whether unused data is consistent with that used to build the model. The iterative nature of the protocol thus extends beyond the generation of the first ‘correct’ model.