 |
|
IMP Tutorial
|
In this example we setup and evaluate a scoring function based on XL-MS data.
First we create the representation using PMI. We create two proteins:
done building "ProtA" Chain A Copy: 0 done building "ProtB" Chain B Copy: 0
To make it easier to see what's going on, we'll place the beads at fixed points in space:
["1-10_bead", "1-10_bead", "11-20_bead", "21-30_bead"]
Next, we'll make some cross-links. The cross-link dataset is a comma separated value (CSV) file with at least the protein and the residue names (no spaces between commas):
Now we create a conversion map between internal keywords of cross-links features and the one in the file:
With this keyword interpreter, let's read the cross-link database:
Let's check that the database looks ok:
1 --- XLUniqueID 1 --- XLUniqueSubIndex 1 --- XLUniqueSubID 1.1 --- Protein1 ProtA --- Protein2 ProtB --- Residue1 1 --- Residue2 10 --- IDScore 1.0 --- Redundancy 1 --- RedundancyList ['1.1'] --- Ambiguity 1 --- Residue1LinksNumber 3 --- Residue2LinksNumber 1 ------------- 2 --- XLUniqueID 2 --- XLUniqueSubIndex 1 --- XLUniqueSubID 2.1 --- Protein1 ProtA --- Protein2 ProtB --- Residue1 1 --- Residue2 11 --- IDScore 2.0 --- Redundancy 1 --- RedundancyList ['2.1'] --- Ambiguity 1 --- Residue1LinksNumber 3 --- Residue2LinksNumber 1 ------------- 3 --- XLUniqueID 3 --- XLUniqueSubIndex 1 --- XLUniqueSubID 3.1 --- Protein1 ProtA --- Protein2 ProtB --- Residue1 1 --- Residue2 21 --- IDScore 2.0 --- Redundancy 1 --- RedundancyList ['3.1'] --- Ambiguity 1 --- Residue1LinksNumber 3 --- Residue2LinksNumber 1 -------------
With the database we can now set up the scoring function. Note the text generated. The program reports the nuisance particles associated to the cross-link (sigma and psi):
gathering copies
defaultdict(<class 'int'>, {'ProtA': 0, 'ProtB': 0})
done pmi2 prelims
generating a new cross-link restraint
--------------
CrossLinkingMassSpectrometryRestraint: generating cross-link restraint between
CrossLinkingMassSpectrometryRestraint: residue 1 of chain ProtA and residue 10 of chain ProtB
CrossLinkingMassSpectrometryRestraint: with sigma1 SIGMA sigma2 SIGMA psi PSI
CrossLinkingMassSpectrometryRestraint: between particles 1-10_bead and 1-10_bead
==========================================
generating a new cross-link restraint
--------------
CrossLinkingMassSpectrometryRestraint: generating cross-link restraint between
CrossLinkingMassSpectrometryRestraint: residue 1 of chain ProtA and residue 11 of chain ProtB
CrossLinkingMassSpectrometryRestraint: with sigma1 SIGMA sigma2 SIGMA psi PSI
CrossLinkingMassSpectrometryRestraint: between particles 1-10_bead and 11-20_bead
==========================================
generating a new cross-link restraint
--------------
CrossLinkingMassSpectrometryRestraint: generating cross-link restraint between
CrossLinkingMassSpectrometryRestraint: residue 1 of chain ProtA and residue 21 of chain ProtB
CrossLinkingMassSpectrometryRestraint: with sigma1 SIGMA sigma2 SIGMA psi PSI
CrossLinkingMassSpectrometryRestraint: between particles 1-10_bead and 21-30_bead
==========================================
We can evaluate this restraint at the current system configuration:
3.0602707946915624
Let's plot the score while moving ProtA bead wrt ProtB. First, we get the particle corresponding to ProtA:
Now we can move ProtA on the x-axis:
We can make a simple plot of the scores against the x coordinate. This plot shows that the system has two minima:
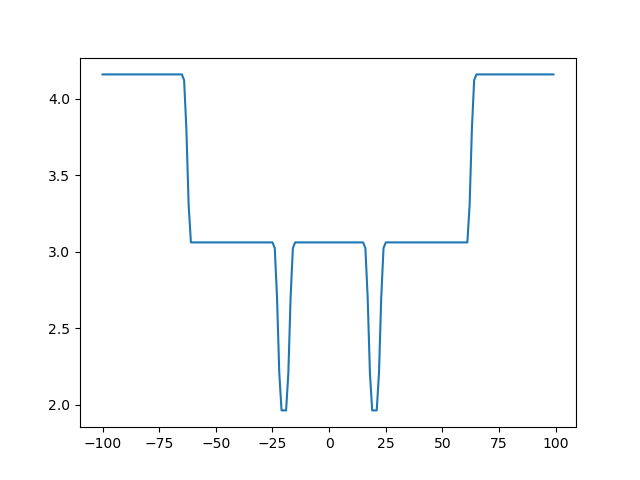
The plot is weird, so let's analyse what is going on.
First let's simplify our dataset, by considering only the first cross-link. Let's filter by the UniqueID, creating a new database that contains only the second cross-link, namely UniqueID=2:
2 --- DataSetName XL --- XLUniqueID 2 --- XLUniqueSubIndex 1 --- XLUniqueSubID 2.1 --- Protein1 ProtA --- Protein2 ProtB --- Residue1 1 --- Residue2 11 --- IDScore 2.0 --- Redundancy 1 --- RedundancyList ['2.1'] --- State 0 --- Sigma1 SIGMA --- Sigma2 SIGMA --- Psi PSI --- Ambiguity 1 --- Residue1LinksNumber 1 --- Residue2LinksNumber 1 --- Particle1 "1-10_bead" Fragment: [1, 11) (99 0 0: 5.9919) --- Particle2 "11-20_bead" Fragment: [11, 21) (0 0 0: 5.9919) --- Particle_sigma1 1e-07 < Scale = 2 < 1000 --- Particle_sigma2 1e-07 < Scale = 2 < 1000 --- Particle_psi 1e-07 < Scale = 0.25 < 0.5 --- Restraint "|XL|2.1|ProtA|1|ProtB|11|0|PSI|" --- IntraRigidBody False --- ShortLabel |XL|2.1|ProtA|1|ProtB|11|0|PSI| -------------
Now we can create a new restraint based on this database and, as before, score while moving ProtA:
gathering copies
defaultdict(<class 'int'>, {'ProtA': 0, 'ProtB': 0})
done pmi2 prelims
generating a new cross-link restraint
--------------
CrossLinkingMassSpectrometryRestraint: generating cross-link restraint between
CrossLinkingMassSpectrometryRestraint: residue 1 of chain ProtA and residue 11 of chain ProtB
CrossLinkingMassSpectrometryRestraint: with sigma1 SIGMA sigma2 SIGMA psi PSI
CrossLinkingMassSpectrometryRestraint: between particles 1-10_bead and 11-20_bead
==========================================
It is clear that the restraint has a minimum when ProtA and ProtB:11-20 are close (namely when ProtA x is around 0). In fact, the restraint has a sigmoid shape:
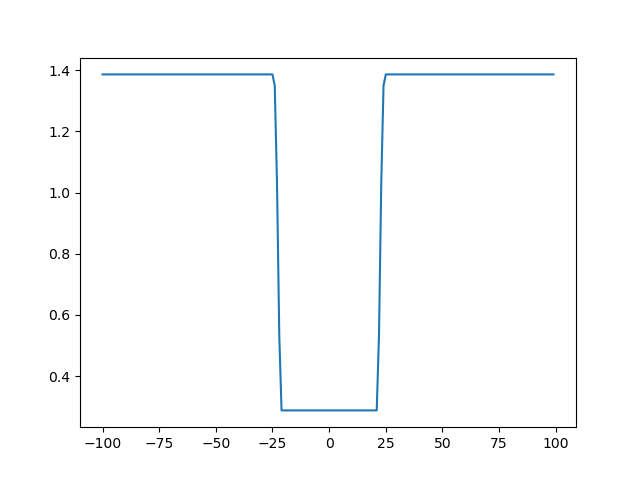
Now let's play with the parameters sigma and psi to understand their roles. Let's get sigma first:
and let's vary its value between 1 and 20 to see what happens:
From the plot, one can see sigma modulates both the slope of the sigmoid and the plateau of the minimum. This is because sigma is the structural uncertainty associated with the position of the cross-linked beads:
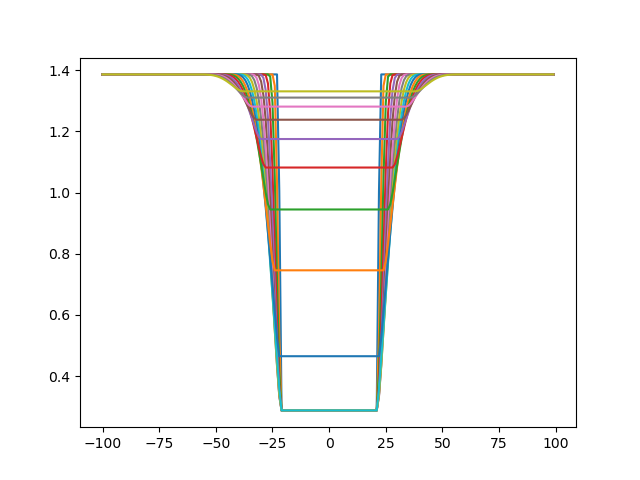
Let's get psi now (setting sigma back to 11), and vary its value between 0.01 and 0.5 to see what happens:
Plotting the values again, one can see psi modulates the plateau of the minimum and the maxima. This is because psi is the uncertainty associated with the cross-link observation:
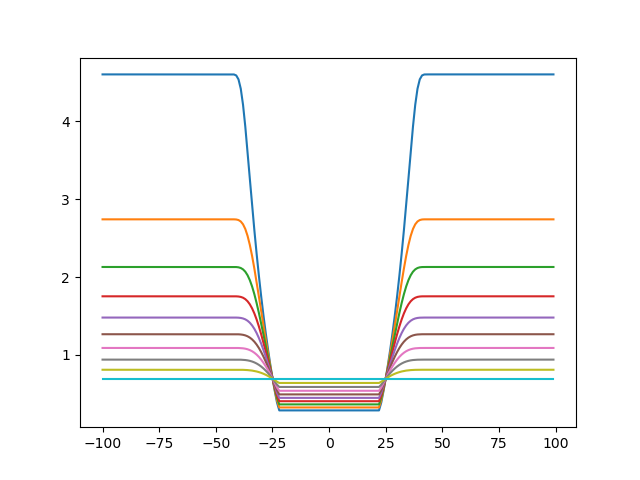
Now let's move the parameters when we consider all three cross-links. First, sigma:
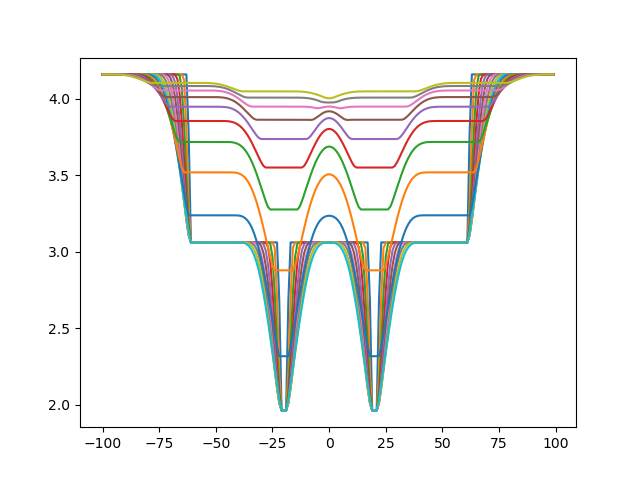
Then, for psi:
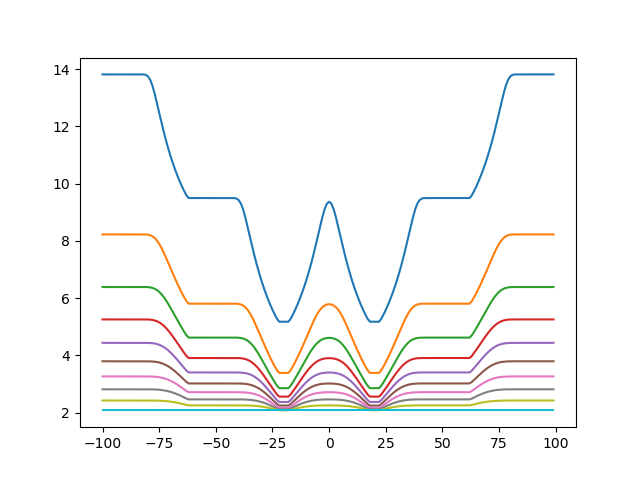
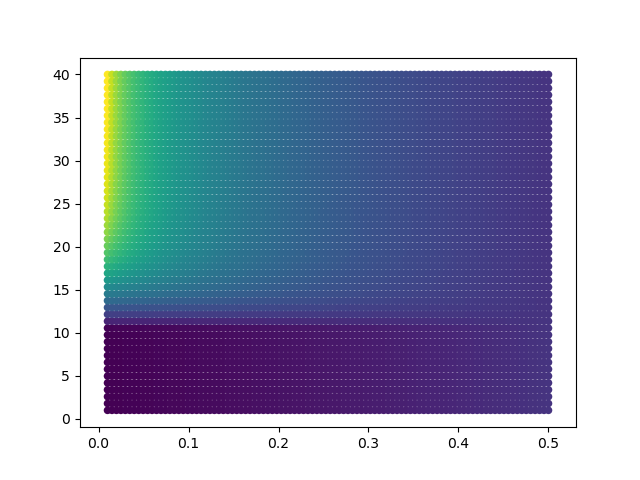
Now we can try to optimize the values of PSI and SIGMA, and see what is the best scoring value, fixing the coordinate of ProtA to a minimum:
There is a minimum when PSI is close to zero and sigma is between 0 and 10:
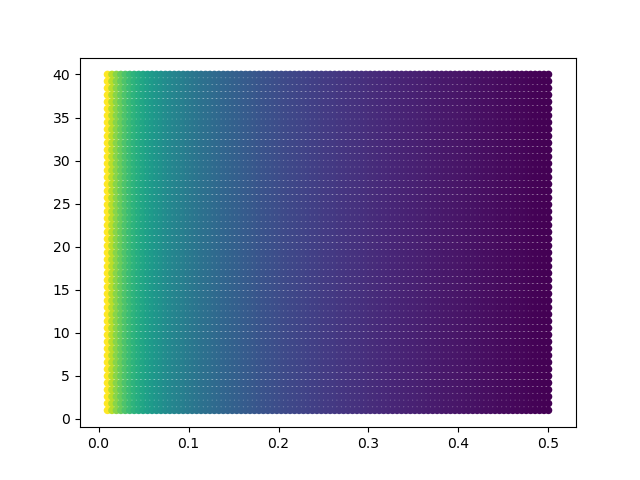
Let's move ProtA away, so that any cross-link is satisfied:
The minimum is at Psi=0.5, irrespective of the value of Sigma:
Ambiguity can also be an issue when dealing with cross-links. See this followup tutorial for further information.
