


 Download files
Download files Verified to work with the
Verified to work with the  To install the software needed to reproduce this system with the
To install the software needed to reproduce this system with the
 To set up the environment on the UCSF Wynton cluster to run
this system, run:
To set up the environment on the UCSF Wynton cluster to run
this system, run:

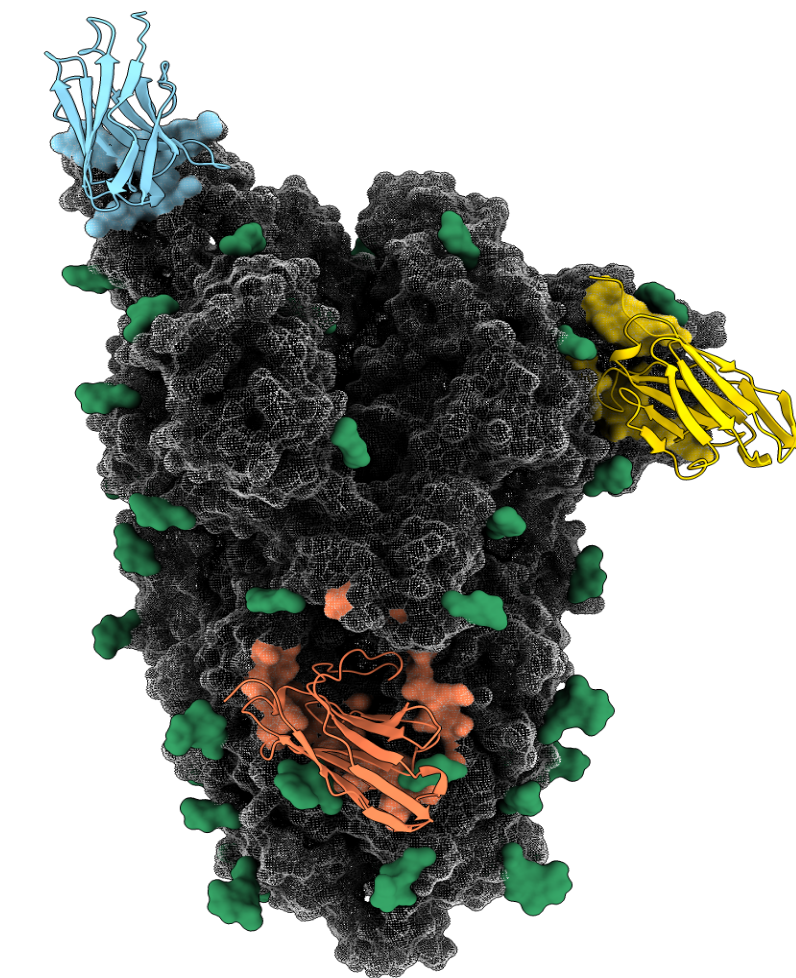
This repository contains comparative models of 21 nanobodies and integrative models of their epitopes on the receptor-binding (RBD) and ectodomains of the SARS-CoV-2 spike protein. Epitopes were modelled using chemical crosslinks and escape mutagenesis data. Both receptor (spike protein) and nanobodies were represented as completely rigid subunits. This work develops a computationally efficient shape complementarity restraint focused around the escape mutant residues, distance restraints between nanobody CDR loops and viral escape residues and a modified interface-metric (inspired by the fcc metric) for clustering alternate models from structural sampling.
Both the receptor and nanobodies in this work have been coarse-grained at a single residue per coarse-grained bead, and subsequently subjected to rigid-rigid docking. Thus the exact orientation of the nanobody on the spike surface maybe noisy and need future refinements. The focus of this exercise is thus to predict a comprehensive epitope on the spike surface that is maximally consistent with input crosslink and escape data.
Nanobody names in this repository are simplified versions of those used in the paper.
| Nanobody name in this repository | Nanobody name in paper | x |
|---|---|---|
| rbd-x | S1-RBD-x | 9, 15, 16, 21, 22, 23, 24, 29, 35, 40 |
| s1-x | S1-x | 1, 6, 23, 36, 37, 46, 48, 49, 62 |
| s2-x | S2-x | 10, 40 |
This is an integrative epitope modeling module written using IMP and PMI. It contains:
restraints: Computationally efficient receptor-ligand shape complementarity restraints optimized for receptor epitopes and antibody-like paratopes (i.e. CDR loops)epitopelibProvides a very fast (cythonized) implementation of the fcc metric, commonly used for comparing interfaces in rigid-rigid docking. The cython file contained here is calledepitopelib.pyx. Please runcd nblib && cythonize -i epitopelib.pyxto produce the.solibrary which can then beimport-ed into python.clustering: Provides anetworkx- based implementation of the Taylor-Butina algorithm for clustering binary interfaces.restraint_satisfaction: Tools for checking satisfaction of restraints used in modeling.graphics: Various tools for converting RMF to PDB files and writing ChimeraX scripts for rendering figures.utils: Miscellaneous utilities used throughout the project.
MODELLER scripts and top scoring comparative models of all 21 nanobodies both before and after loop refinement. All 21 nanobodies are modelled from the human Vsig4 targeting nanobody Nb119.
Contains nanobody structures generated by AlphaFold-2. The ColabFold notebook framework was used to run AlphaFold, specifically the AlphaFold2_batch Colab notebook with default settings, and structure relaxation post prediction. For each nanobody, the top ranked nanobody according to average plddt was selected for epitope modeling.
Contains scripts that use the nblib module to structurally sample, cluster and calculate restraint satisfaction for nanobody binding modes on the spike protein. Sub-folders:
-
scripts: Python scripts for sampling binding models and subsequent analysis. -
wynton_job_scripts: SGE scripts used to run structural sampling on the UCSF Wynton compute cluster. -
data: Contains sequences of RBD, ectodomain and all modeled nanobodies, cryo-EM structures of spike RBD and ectodomain and comparative models of nanobodies (data/pdb), PMI topology files (data/topology), crosslinks (data/xl) list of viral escape mutants and the CDR regions of the corresponding nanobodies (data/escape_mutant), and configuration (json) files for modeling each binary receptor-nanobody complex that contains multiplicative weights for the different restraints used in modeling (data/config). -
results: Contains PDB structures of 21 nanobodies on different parts of the spike. In each of these files, the nanobody is chain "A" and the spike is chain "0" (RBD monomer) or chains "1", "2" and "3" (ectodomain trimer). Structural sampling and clustering produces rigidly-docked binary complexes in the RMF file format. Subsequently an utility innblib.graphicsconverts the RMF to a PDB file. Binary complex pdb files are named as [receptor]_[ligand].pdb where receptor is one of rbd, ntd or ectodomain and ligand names are as column 1 in the table above.results/chimerax_renderingcontains ChimeraX scripts for rendering all nanobody epitopes simultaneously on the spike surface. Used to generate Fig 6 in the paper.
The overall content is similar as above. Main differences are -
- integrative modeling and analysis scripts now have additional command line flags for selecting SARS-CoV-2 variant (Wuhan, Delta, Omicron BA.1, BA.4, XBB.1, BQ.1.1)
- results contain scripts to heat-map relative binding (Kd) or potency (IC50) data on consensus epitopes of nanobodies belonging to the same group.
Author(s): Tanmoy Sanyal
Date: December 4, 2021
License: CC BY-SA 4.0 This work is licensed under the Creative Commons Attribution-ShareAlike 4.0 International License.
Last known good IMP version:
Testable: Yes.
Parallelizable: Yes
Publications:
-
Fred D. Mast, Peter C. Fridy, Natalia E. Ketaren, Junjie Wang, Erica Y. Jacobs, Jean Paul Olivier, Tanmoy Sanyal, Kelly R. Molloy, Fabian Schmidt, Magdalena Rutkowska, Yiska Weisblum, Lucille M. Rich, Elizabeth R. Vanderwall, Nicholas Dambrauskas, Vladimir Vigdorovich, Sarah Keegan, Jacob B Jiler, Milana E. Stein, Paul Dominic B. Olinares, Louis Herlands, Theodora Hatziioannou, D. Noah Sather, Jason S. Debley, David Fenyo, Andrej Sali, Paul D. Bieniasz, John D. Aitchison, Brian T. Chait, Michael P. Rout, Highly synergistic combinations of nanobodies that target SARS-CoV-2 and are resistant to escape, eLife 2021; 10e73027
-
Natalia E. Ketaren, Fred D. Mast, Peter C. Fridy, Jean Paul Olivier, Tanmoy Sanyal, Andrej Sali, Brian T. Chait, Michael P. Rout, John D. Aitchison, Nanobody repertoire generated against the spike protein of ancestral SARS-CoV-2 remains efficacious against the rapidly evolving virus, Biorxiv 2023