 |
|
IMP Manual
for IMP version 2.21.0
|
In this example, MultiFit is used to build a model of porcine mitochondrial respiratory complex II (PDB id 3sfd), using crystal structures of its 4 constituent proteins, a cryo-electron microscopy (EM) map of the entire complex, and information from proteomics.
All steps in the procedure use a command line tool called multifit. For full help on this tool, run from a command line:
First, obtain the input files used in this example and put them in the current directory, by typing:
(On a Windows machine, use copy rather than cp.) Here, <imp_example_path> is the directory containing the IMP example files. The full path to the files can be determined by running in a Python interpreter 'import IMP.multifit; print(IMP.multifit.get_example_path('3sfd'))'.
The first step is to create an input file listing the subunits involved in the complex. This file is a text file with a simple format; it simply contains one line per component with the following information: the name that MultiFit will use for the component, the name of the file containing the atomic coordinates, and flag indicating whether placements of the subunit should be sampled locally (0) or globally (1). The default for the fitting flag is 1 (global search). If the user has prior knowledge or a good hypothesis as to the subunit position, he or she can provide the proposed subunit placement in the atomic coordinates file and ask for a local search.
In this case, no prior knowledge is assumed, and so the subunits file looks like:
3sfdA 3sfd.A.pdb 1 3sfdB 3sfd.B.pdb 1 3sfdC 3sfd.C.pdb 1 3sfdD 3sfd.D.pdb 1
This file is already provided, as 3sfd.subunits.txt.
The next step is to create two other input files that guide the MultiFit protocol. This is done by using MultiFit's 'param' command, by running on a command line:
The 'param' command takes as arguments the coarseness level in residues (30), the resolution of the map in angstroms (15), the map spacing in angstroms (3), the density threshold (335), and the origin of the map in angstroms (27.0, -6.0, 21.0). The spacing (or pixel size) and origin are often stored in the map header. To view the map header, run:
The resolution is typically not stored in the map header; it is usually provided in the corresponding publication and can also be found in the corresponding EMDB entry. A threshold is often provided by the author in the EMDB entry as "Recommended counter level" under the "Map Information" section. Alternatively, IMP provides a utility to calculate an approximate counter level based on the molecular mass of the complex, which can be run as:
The first file generated by MultiFit, 3sfd.asmb.input, provides information on each of the subunits and their assembly density map, such as names of the files from which the input structures and map will be read, and those to which outputs from later steps will be written. The second file, 3sfd.asmb.alignment.param, specifies scoring and optimization parameters for each step of the MultiFit protocol. These parameters can be adjusted if necessary to handle difficult modeling cases.
(Note that two other files are also created, with a .refined extension. These can be used for model refinement, which is discussed later.)
Next, a reduced representation of the assembly density map is generated using the Gaussian Mixture Model, by running:
This command computes a reduced representation of the EM map that best reproduces the configuration of all voxels with density above the density threshold (provided in the 3sfd.asmb.input file) as a set of 3D Gaussian functions. (The default number of Gaussians is the number of components. However, if the sizes of the subunits differ, it is recommended to use the -s option to set the number of residues encapsulated in each Gaussian. For example, with 50 residues per Gaussian, a 170-residue protein should use 3 Gaussians and a 260-residue protein should use 5 Gaussians.) The reduced representation is written out as a PDB file containing fake CA atoms, where each CA corresponds to a single anchor point, and also as a Chimera cmm file.
First, fit each protein to the map using a FFT search either globally or locally:
The output is a set of candidate fits. In each file, a single subunit is rigidly rotated and translated to fit into the density map. Each fit is written out as the transformation (rotation and translation) required to place the original subunit in the density map. The fitting of a subunit into the density map is performed by globally searching for subunit transformations yielding high cross-correlation between the subunit and the map via a fast Fourier transform.
Next, a list of valid fit indexes is created. As below, this list is simply the top 3 hits from fit_fft, but they could be filtered by other criteria (e.g., proximity to anchor points) if desired. Note that the more fit indexes used, the longer it will take to combine the fits into a global solution in further steps in the protocol (but the more likely it is that the optimum solution is found). For a quick demonstration, the 3 top fits are sufficient, but 10 or more fits are recommended in most cases. Do this by running:
Here, the restraint file used in the next assembly step is created. This file instructs MultiFit how to combine the individual subunit fits created above into a global solution of all subunits simultaneously fitted into the map. First, MultiFit can generate a basic proteomics file, indicating between which pairs of proteins a complementarity restraint (i.e., that the surfaces of the proteins should fit and complement each other) should be calculated:
The user can then add additional information from proteomics experiments to this file. Here, 7 simulated residue-residue cross-link restraints are added. The excluded volume (EV) pairs are also updated to calculate complementarity restraints between pairs of proteins as indicated by the cross-link restraints. After these additions, the final 3sfd.asmb.proteomics file looks like:
|proteins| |3sfdA|1|613|nn|nn| |3sfdB|1|239|nn|nn| |3sfdC|1|138|nn|nn| |3sfdD|1|102|nn|nn| |interactions| |residue-xlink| |1|3sfdB|23|3sfdA|456|30| |1|3sfdB|241|3sfdC|112|30| |1|3sfdB|205|3sfdD|37|30| |1|3sfdB|177|3sfdD|99|30| |1|3sfdC|95|3sfdD|132|30| |1|3sfdC|9|3sfdD|37|30| |1|3sfdC|78|3sfdD|128|30| |ev-pairs| |3sfdB|3sfdA| |3sfdB|3sfdC| |3sfdC|3sfdD|
This modified file is already present, as 3sfd.xlinks.proteomics. Replace the basic file with it by running:
Note that these restraints will be used to create DOMINO’s junction tree. DOMINO works most efficiently if the size of the intermediate subsets is small. Use the multifit merge_tree command to view the tree defined by the restraints. To reduce the size of the subsets, the user can determine which restraints are used to define the merge tree by setting the first value in the xlink definition. Setting the value to 0 instead of the default 1 specifies that the restraint is evaluated only at the root of the tree and not in an intermediate merging step.
The fits are combined into a set of the best-scoring global configurations by running:
The output is the file 3sfd.asmb.combinations which contains a ranked list of combinations (best scored first). Each combination is simply a list of 4 numbers, where the first number is the index of the fit for the first subunit, the second number the fit for the second subunit, and so on.
The scoring function used to assess each fit includes the quality-of-fit of each subunit in the map, the protrusion of each subunit out of the map envelope, the shape complementarity between subunits, as indicated in the proteomics file, and distance restraints as defined by proteomics data, also from the proteomics file. The optimization avoids exhaustive enumeration of all possible mappings of subunits to anchor points by means of a branch-and-bound algorithm combined with the DOMINO divide-and-conquer message-passing optimizer using a discrete sampling space.
Finally, models can be generated as PDB files from the fits and best combinations by running:
This generates PDB files for the 5 best-scoring solutions, calling them model.0.pdb, model.1.pdb, and so on.
The best-scoring solution, shown fitted in the density map of the complex, is shown below:
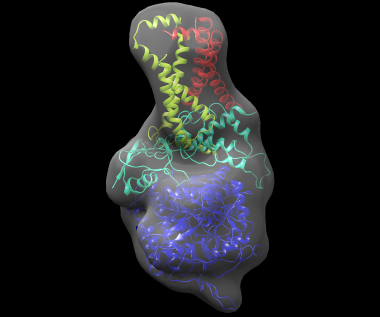
If a reference structure for each subunit is available, the 'reference' command can be used to compare the models to the reference. To use this command, modify 3sfd.asmb.input and add the filename of each reference subunit structure in the rightmost column. In fact, the input subunit structures are already in their native conformation, so these structures can be used. The file 3sfd.asmb.input.ref already has this modification. Then run:
This will report, for the top 5 combinations, the all-atom RMSD between the model and the reference structure of the complex, and a list of placement scores, one for each subunit. Each placement score is the distance that the subunit must be moved to reach the reference structure and the angle through which it must be rotated.
It is often useful to cluster the solutions. MultiFit includes a 'cluster' command to accomplish this. It uses the fastcluster Python module, which needs to be downloaded and installed separately.
To cluster the solutions previously obtained, run:
This calculates the distance between each pair of solutions and makes clusters where no two structures are separated by an RMSD of more than 2 angstroms. In this case, this results in three equally-sized clusters. It generates a file 3sfd.asmb.combinations.clustered containing the combination of a representative for each cluster.
Once the set of models has been determined, they can be improved by refinement. The first step in refinement is to run another FFT fit. This time the fit is run locally around the protein placements as determined by the first run of MultiFit, rather than globally:
In this case, the best-scoring combination (the first one, index 0) is used as the starting point. This generates a new set of subunit fits, with a .refined file extension. These can then be assembled with DOMINO into global solutions, in exactly the same way as before. First, indexes are generated:
Then, the combinations are assembled with DOMINO (note that this uses the 'refined' parameter file generated above):
Note that MultiFit always treats each subunit rigidly. However, improved fits may require the protein to be treated flexibly, so that it can deform to more closely fit in the density. Such flexible fitting is not performed by MultiFit, but the MultiFit-generated models can be passed to a flexible fitting program for further refinement. Two such programs are Flex-EM and MDFF.


