|
|
IMP Manual
for IMP version 2.12.0
|
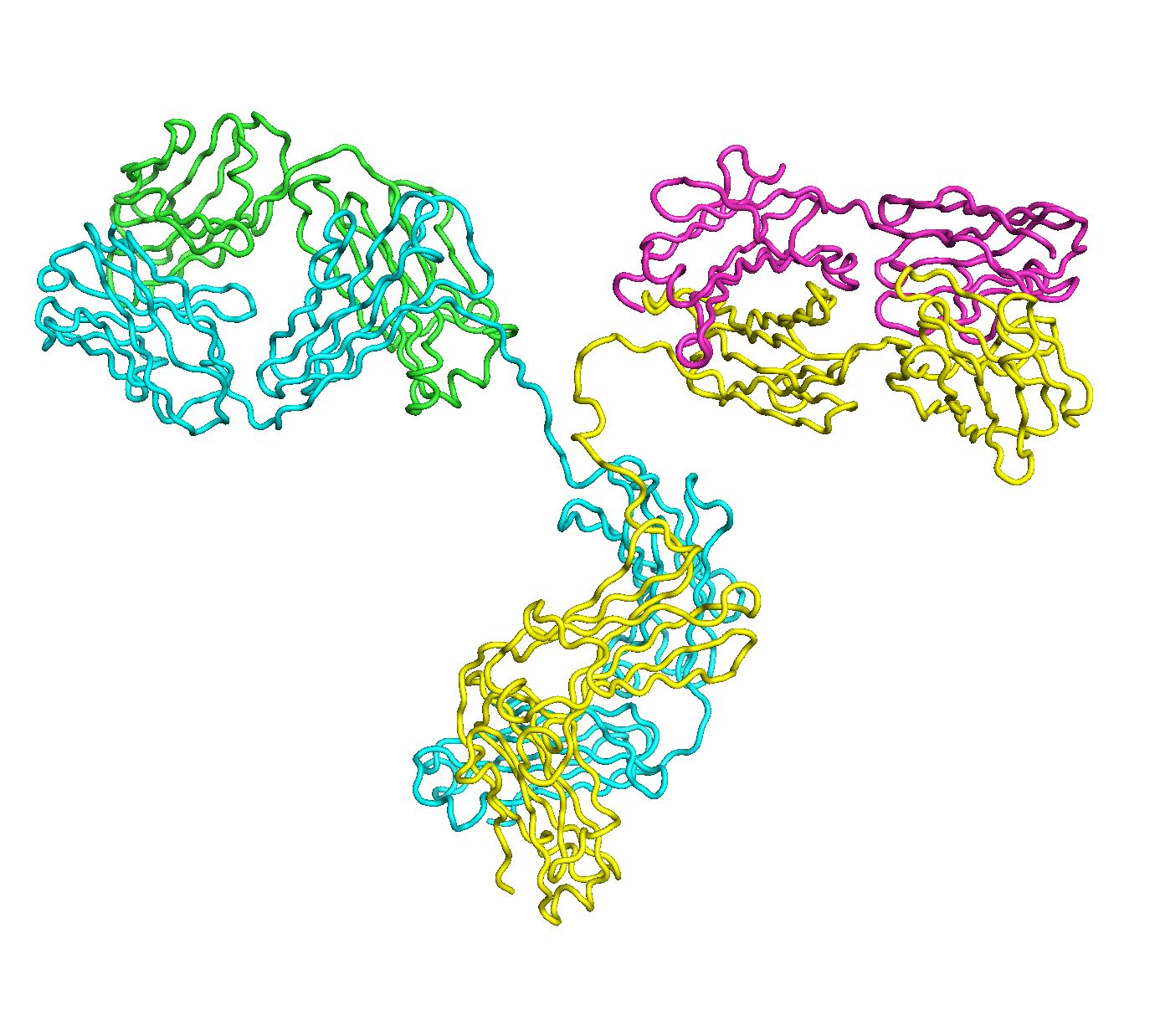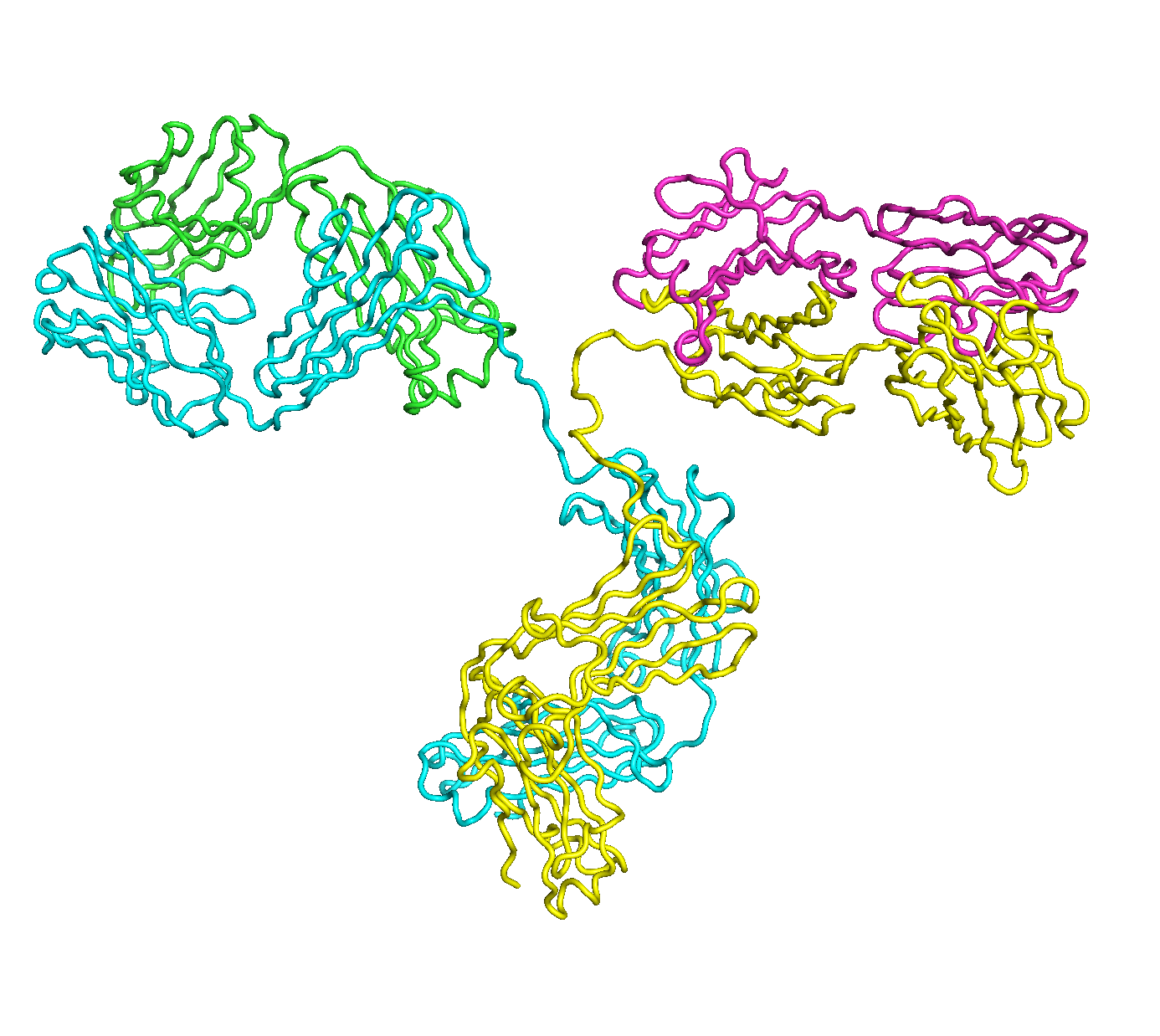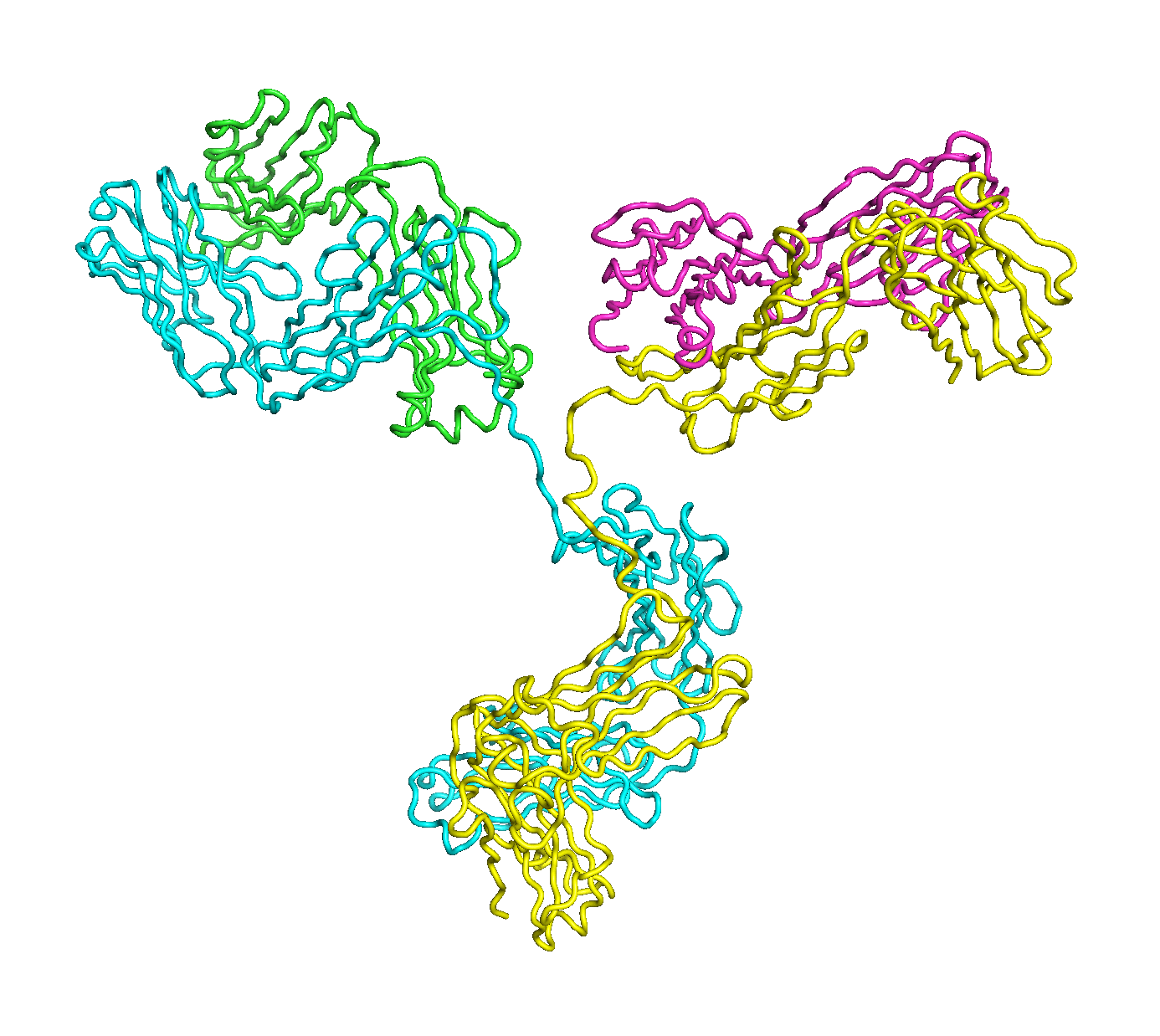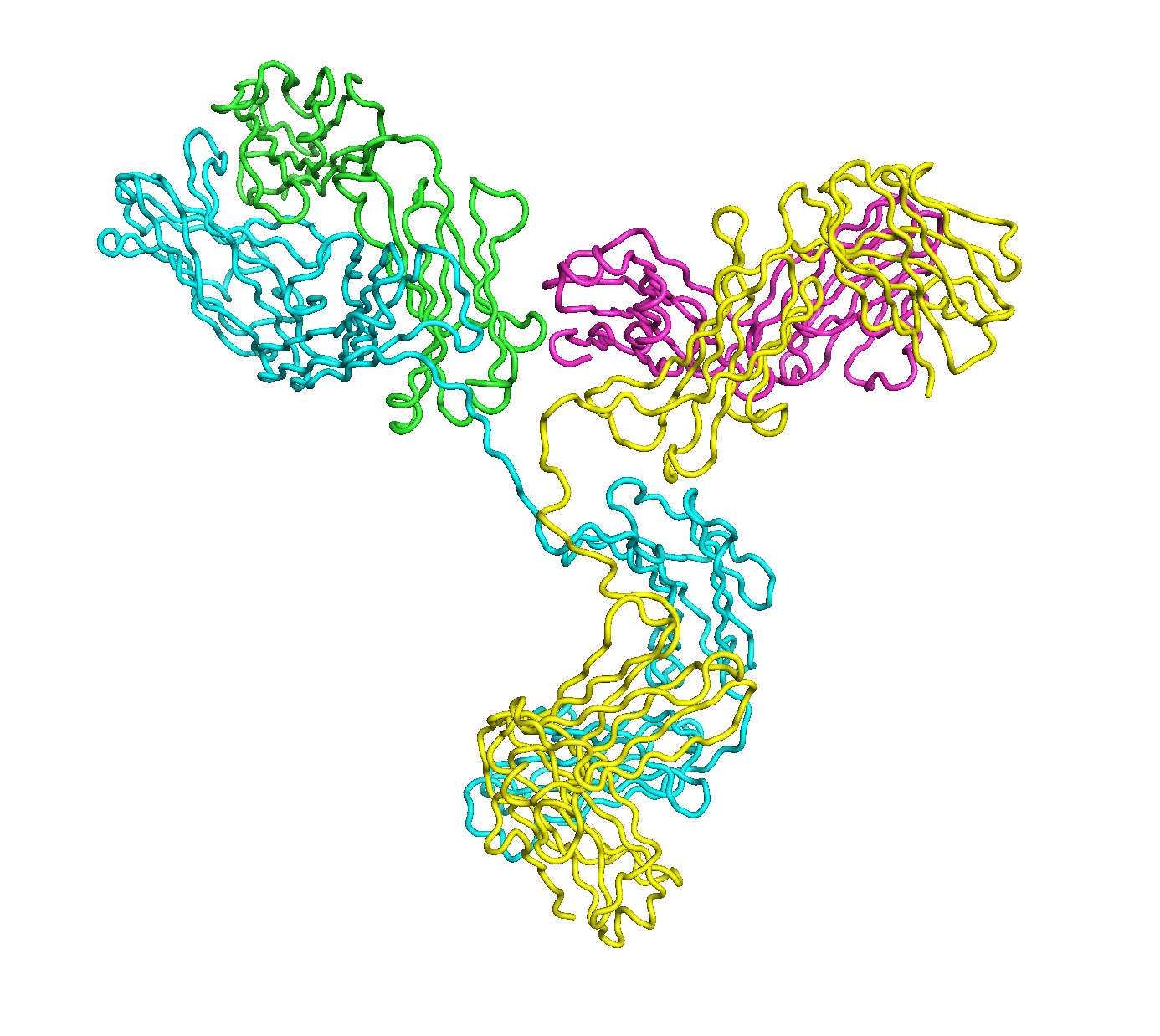
A full length antibody structure consists of two Fab domains and an Fc domain. There are hinge regions between the Fc and Fab domains that we sample here to explore the range of conformations accessible to the antibody.
IMP includes a command line tool rrt_sample for running rapidly exploring random tree (RRT) based sampling, which will be demonstrated in this example.
For full help on the rrt_sample tool, run from a command line:
First, obtain the input files used in this example and put them in the current directory, by typing:
(On a Windows machine, use copy rather than cp.) Here, <imp_example_path> is the directory containing the IMP example files. The full path to the files can be determined by running in a Python interpreter 'import IMP.kinematics; print(IMP.kinematics.get_example_path('antibody'))'.
The structure of the antibody is available in the RCSB Protein Data Bank (PDB) as code 1igt (file 1igt.pdb). The atomic structure can be sampled by running rrt_sample. First, we need to define the linker regions (residues 229-235 in chains B and D). These residues are listed in the input file linker.txt. Since the antibody contains four chains (A-D), we would like to connect them into a single rigid body using '-c' connect_chains option. We do that for the antibody structure using the S-S bonds between the chains. Therefore, the file conect_chains.txt specifies atom numbers for the two chains we want to connect.
By default the method will run RRT for 100 iterations or until it generates 100 conformations. To generate more use -i 1000000 -n 10000 options.
The output files are PDB files with each conformation as a MODEL. By default the program writes 100 conformations into each file. This can be changed using the -m option.